Data Integration
Drag file(s) here to upload.
Alternatively, you can select files by clicking here
Drag file(s) here to upload.
Alternatively, you can select files by clicking here
Integration Results
Some text in the Modal..
×Integration Help
Integration refers to the process of combining data from multiple sources or experiments to create a unified dataset. These sources could be different individuals, biological conditions, batches, technologies (e.g., different sequencing platforms), or tissues. Integration is crucial because it addresses the variability and technical noise that often arises when data is collected from separate experiments or labs, making it possible to compare and analyze the data as a whole.
When performing scRNA-seq experiments across multiple conditions or batches, technical and biological factors can introduce unwanted variations into the data. This variation, often referred to as "batch effects" or "technical noise," can obscure the true biological signals you're interested in. Integration helps to account for these discrepancies, ensuring that cells from different sources can be compared more accurately.
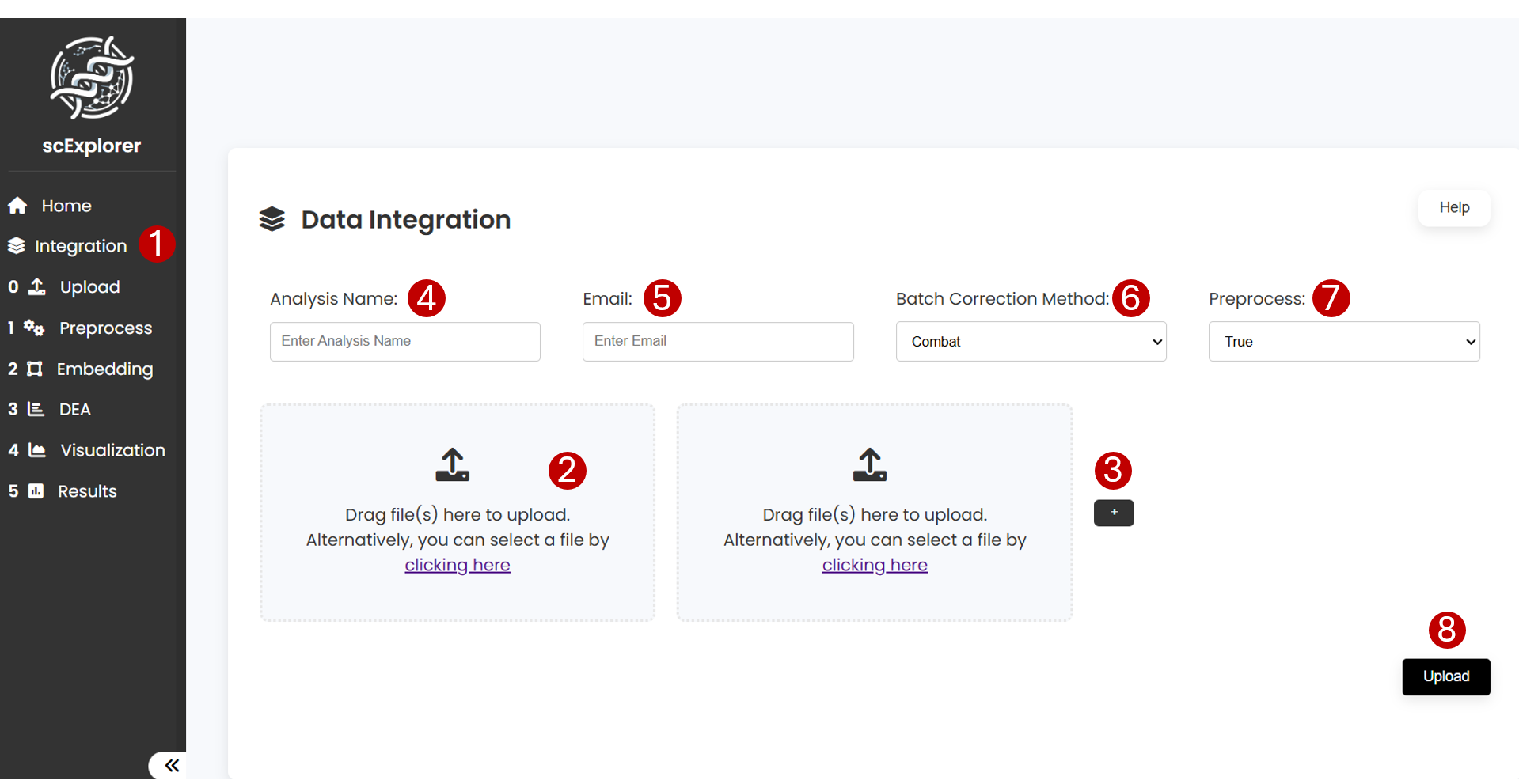
To integrate your data with scExplorer go to Integration (1) section. Next upload or drag individual files in (2). If more than two datasets need to be integrated, click in (3). Next, indicate an Analysis Name (3), and Email (4) to notify when the analysis has finished, the Batch Correction Method to use during the integration (5), and indicate whether the dataset is Preprocessed (6). Finally, click on Upload (7) button to start the analysis.
scExplorer offers four different batch correction methods: Combat, Scanorama, BBKNN, and Harmony.
Combat is a statistical method originally developed for correcting batch effects in microarray data but has been adapted for single-cell RNA-seq data. It adjusts for known batch effects by applying an empirical Bayes framework.
Scanorama is a graph-based method designed specifically for single-cell RNA-seq data integration. It aligns multiple datasets by identifying mutual nearest neighbors (MNNs) between them, then stitches the datasets together based on these commonalities.
BBKNN (Batch Balanced K-Nearest Neighbors) is a KNN-based batch correction method that operates in the context of UMAP or other dimensionality reduction techniques. It works by adjusting the neighbor search to be batch-aware, ensuring that each cell's neighbors come from different batches.
Harmony is a fast and scalable batch correction method designed to handle complex multi-dataset integrations. It iteratively adjusts the cell embeddings in reduced-dimensional space to align cells across batches while preserving biological variation.
Each one of these methods have advantages and disadvantages. Thus, we encourage users to explore all of these methods.
Currently, scExplorer can handle the integration of up to 10 datasets simultaneously in this integration stage. This limitation ensures optimal performance and reliable results for your batch correction analysis.
